The U.S. Food and Drug Administration approved a new drug on Friday that reduces the complications associated with sickle cell disease — the first drug approved for the blood disorder in more than 20 years.
The drug, called Endari, consists of L-glutamine, which is an amino acid, and is approved for sickle cell patients five years and older.
“I am hoping we are finally seeing channels opening, and that this will be the first of many new drugs to hit the market [for sickle cell disease],” said Dr. Alexis A. Thompson, head of the Hematology Section and Director of the Comprehensive Thalassemia Program at the Ann and Robert H. Lurie Children’s Hospital of Chicago.
Sickle cell disease, which predominately affects African-Americans, Latinos and other minority groups, is an inherited lifelong disorder in which red blood cells that are normally round and disc-shaped are instead crescent or sickle-shaped due to abnormal hemoglobin.
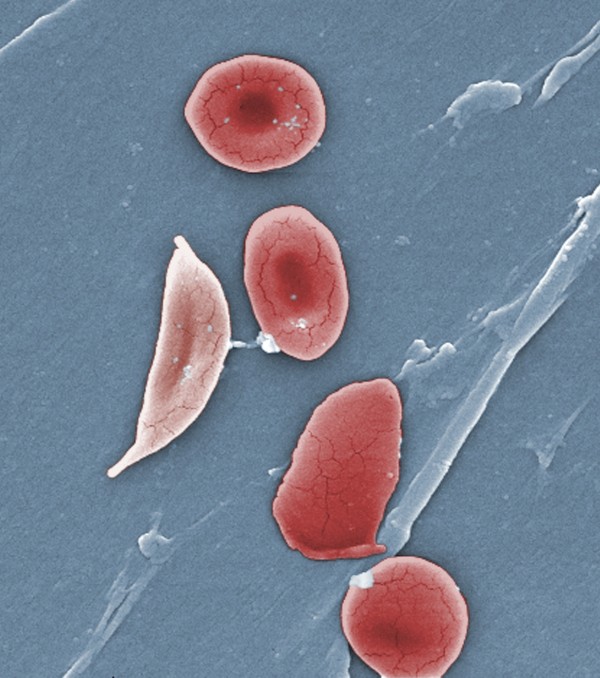
These sickle-shaped cells are not flexible, and they can stick to the walls of blood vessels, causing blockages that stops the flow of blood to organs or tissues. This is the hallmark of the painful, sometimes debilitating, sickle cell pain crises which often require an Emergency Department visit or hospitalization. The crises can occur without warning or can be triggered by stressors such as cold weather, dehydration or pregnancy.
“These [crises] are major disruptions to the individuals who have sickle cell disease,” Thompson said. “Not just the severity of pain, but their ability to continue with their education and to maintain their family life and a job. All are impacted if one has to be hospitalized frequently for something like pain.”
Sickle cell disease affects 100,000 Americans, and the estimated annual costs of medical care in the United States for the disease exceeds $1.1 billion according to the a 2009 estimate, Reuters reported.
Persons with sickle cell disease inherit two abnormal hemoglobin genes, one from each parent. One in 13 African-American babies is born as a genetic carrier of sickle cell, called sickle cell trait, and one in every 365 African-American children is born with sickle cell disease, according to the National Heart, Lung and Blood Institute. As a result, every state and the District of Columbia requires that every baby is screened for sickle cell disease or sickle cell trait.
“We are very effective in identifying newborns [with sickle cell disease] in the U.S,” says Thompson. “We are able to provide vaccinations, penicillin, close medical management. But, when the complications arise, we have very few tools specific to sickle cell to offer patients.”
That’s what makes Endari exciting, she says.
In sickle cell disease, the red cells do not have enough of the amino acid L-glutamine, which makes the cells more fragile, and more prone to clog blood vessels. Endari works by increasing the levels of L-glutamine in the blood.
Endari was studied in patients ages five to 58 years old, and researchers found that the patients who received the medication had fewer hospitalizations and shorter hospitalizations than those who took the placebo.
Like with any medication, there are side effects. The FDA reports that common side effects of Endari include constipation, nausea, headache, abdominal pain, cough, pain in the extremities, back pain and chest pain.
“We clearly have much room for improvement in what we can provide for people with sickle cell disease and while we are clearly very excited to see another drug reach the marketplace, we need more,” Thompson said.
[“Source-nbcnews”]