Scientists have identified a key mechanism that damages the cornea in Meesmann epithelial corneal dystrophy, a rare blinding disorder, which is passed from parents to their children.
Researchers at the Universities of Dundee and Ulster, working with colleagues in Denmark, have discovered that faults in a gene that is responsible for corneal cell structure and stability lead to protein misfolding and cell death.
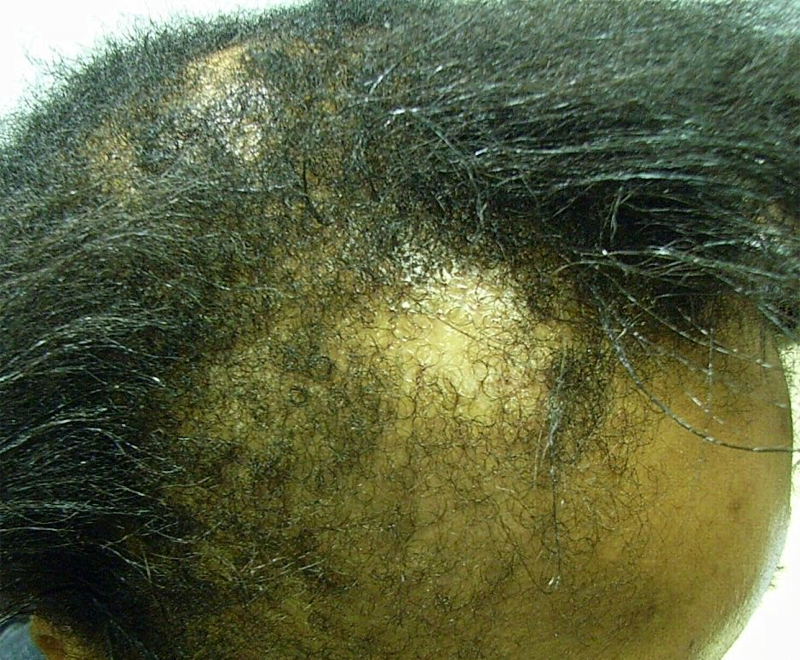
Meesmann epithelial corneal dystrophy (MECD) is a rare inherited condition in which very small cysts (microcysts) form in the outer layer (epithelium) of the cornea. Symptoms include sensitivity to light (photophobia), blurred vision, foreign-body sensation and ruptured cysts. In its most severe form MECD leads to scarring and clouding of the central cornea and can result in blindness.
The same process examined by the researchers is also active in Alzheimer’s, cystic fibrosis and other eye disorders such as retinitis pigmentosa.
The research has been funded in the UK by Fight for Sight, Wellcome Trust and the Medical Research Council.
Previous research has identified 23 faults in the gene KRT12 and three in KRT3 that cause some degree of MECD. These genes encode keratins K12 and K3, respectively; cornea-specific proteins that bind together to form the internal scaffolding of cells in the cornea’s epithelial layer.
However, the means by which faulty K12 leads to microcysts forming and bursting was poorly understood, with research progress hindered by the lack of an appropriate animal model of MECD.
In the current study, published in the journal Human Molecular Genetics, the researchers report the first mouse model of MECD that replicates the human condition. Mice were engineered to carry two copies of the human version of a fault known as K12-Leu132Pro which, in humans, results in the most severe form of MECD.
The team studied the structure of the cornea and the distribution of keratin in the K12 mice. The results were then compared to human tissue samples from the cornea.
Results showed that the corneal epithelium of the K12 mice was 50% thicker than in normal mice, with a disorganised cell structure and signs of cell fragility. There were also changes to the pattern of gene and protein activity in the cornea, with a set of keratin genes that were more or less active than normal and increased activity of a protein called CHOP, which is involved in cleaning up misfolded protein from cells. A similar pattern of changes was seen in the human tissue samples from a patient with the same K12-Leu132Pro fault, compared to healthy human corneal tissue.
Professor Irwin McLean, Professor of Human Genetics in the College of Life Sciences at the University of Dundee, said:”We knew that protein misfolding was likely to play a role in Meesmann epithelial corneal dystrophy because of the critical location of the K12 fault but this is the first direct evidence of it. What we see in the K12 mouse is the protein signature of the unfolded protein response – a pathway that’s activated to clean up debris from misfolded protein. However prolonged activation of this pathway can trigger programmed cell death, and we think this is happening in both mouse and human cornea as the K12-Leu132Pro fault produces overwhelming keratin misfolding.”
Professor Tara Moore, group leader for vision science research in the School of Biomedical Science at Ulster University, said:”The apparent activation of the unfolded protein response in MECD adds to the evidence that the eye is highly susceptible to cellular stress, as we see in other disorders such as keratoconus, cataract and diabetic retinopathy. However we still need to establish the specific pathway involved. Our K12 model will prove useful both here and in future as a test bed for therapies that target corneal stress responses and other keratin disorders caused by similar genetic faults.”
Dr Dolores M Conroy, Director of Research at Fight for Sight, said: “The top priority for corneal research identified by the Sight Loss and Vision Priority Setting partnership was to answer the question of whether gene or stem cell therapies can be developed. Identifying the means by which a genetic fault causes harm is a vital step on that path. The humanised mouse model of Meesmann Corneal Dystrophy developed in this study will be invaluable for further understanding of the disease mechanisms and for the testing of new therapies that could prevent protein misfolding, or gene editing to correct the genetic fault.”
[Source:- Medicalnewstoday]